

Médecine interne
Chapitre S03-P01-C27
Maladie associée aux IgG4
et Nicolas Schleinitz
| ATTENTION : Les informations contenues dans ce chapitre sont susceptibles d’être obsolètes, il existe une version plus récente de ce chapitre. |
| Lien vers la mise à jour |
Le terme de maladie associée aux immunoglobulines G4 (MAG-4) est d’introduction très récente [8]. Il correspond à la traduction de la terminologie anglo-saxonne IgG4-related disease retenue lors du premier symposium international sur cette maladie, en octobre 2011. Il doit permettre d’éviter la confusion, liée aux nombreuses dénominations différentes de cette maladie, utilisées ces dernières années dans la littérature médicale [9]. À côté de ce terme général qui englobe les différentes manifestations de la maladie, il a été proposé de renommer les différentes atteintes d’organes en y associant le suffixe « associé(e) aux IgG4 ». De nombreux syndromes correspondant à des manifestations de la maladie associée aux IgG4 ont été ainsi renommés. Par exemple, le syndrome de Mikulicz devient la sialadénite et/ou la dacryo-adénite associée aux IgG4 et la pancréatite sclérosante ou auto-immune de type 1 devient la pancréatite associée aux IgG4 [8]. La maladie associée aux IgG4 recouvre ainsi différentes atteintes d’organes, souvent associées chez un même patient, qui ont en commun des caractéristiques cliniques, biologiques et histologiques particulières. Elle touche plus volontiers les hommes et a un profil évolutif chronique marqué par des rechutes fréquentes. L’évolution fibrosante peut être responsable de séquelles. Sa physiopathologie reste encore largement méconnue. Le traitement de première ligne repose actuellement sur la corticothérapie. La fréquence élevée des rechutes et l’existence de formes réfractaires expliquent l’utilisation fréquente chez ces patients de traitements immunosuppresseurs en seconde ligne.
Historique
Le premier lien à avoir été fait entre l’élévation des IgG4 sériques et une pathologie inflammatoire polyclonale et fibrosante date de 2001 [5] concerne la pancréatite dite « auto-immune » de type 1, ou sclérosante, qui reste, dans les séries actuelles, la manifestation la plus fréquente de la maladie. Le concept de pancréatite sclérosante a été proposé dès 1961 par des gastro-entérologues français, devant l’association chez certains patients d’une pancréatite fibrosante d’évolution chronique, idiopathique, à une hypergammaglobulinémie polyclonale. En 1991, deux observations de pancréas pseudo-tumoraux, dont l’étude histologique retrouve un infiltrat lymphoplasmocytaire dense avec fibrose interstitielle abondante et des images de phlébites oblitérantes, sont rapportées par une équipe japonaise. Les mêmes anomalies histologiques sont rapportées au niveau des voies biliaires, et le terme de cholangite et de pancréatite lymphoplasmocytaire sclérosante est proposé. En 1995, le terme de pancréatite « auto-immune » est avancé. Il repose sur l’association d’une pancréatite associée à une hypergamma-globulinémie polyclonale et une sensibilité aux corticostéroïdes. L’élévation significative des IgG4 sériques chez les patients atteints de pancréatites sclérosantes en comparaison aux patients avec cancer du pancréas, pancréatite chronique calcifiante ou syndrome de Gougerot-Sjögren est rapportée en 2001 [5]. C’est la même équipe qui décrit un an plus tard la présence de nombreux plasmocytes IgG4+ dans les tissus de biopsies de fibroses rétropéritonéales et de pancréatites de trois patients présentant des IgG4 sériques élevées. Ils concluent à l’association des pancréatites sclérosantes avec une pathologie systémique sclérosante et à un rôle potentiel des IgG4 dans la physiopathologie de cette maladie. À partir de ces deux observations, l’étude « systématique » du taux sérique des IgG4 et de la présence de plasmocytes IgG4+ en nombre important dans des prélèvements tissulaires va amener à regrouper différents syndromes ou entités dans le cadre de la maladie associée aux IgG4. C’est le cas du syndrome de Mikulicz, de certaines fibroses rétropéritonéales « idiopathiques », de certaines néphrites interstitielles ou encore d’aortites inflammatoires.
Physiopathologie
La physiopathologie de la MAG-4 est encore largement méconnue. Différentes hypothèses peuvent être formulées à partir des rares données épidémiologiques et des lésions élémentaires retrouvées en histologie conventionnelle et en immunohistologie. Il s’agit d’une pathologie acquise survenant principalement au cours des cinquième et sixième décennies chez l’homme [2], [9]. Les cas pédiatriques semblent exceptionnels. Il existe une association avec l’allergie [2], variable selon les séries, une élévation fréquente des IgE totales et la présence fréquente d’éosinophiles au sein des lésions tissulaires. Ces données suggèrent une orientation de la réponse immunitaire de type TH2, qui est d’ailleurs documentée [4bis]. En effet, le profil de production de cytokines au niveau tissulaire est de type TH2, avec une augmentation d’expression des ARNm ou, en immunohistochimie, des interleukines IL-4, IL-5, IL-10, CCR4 (chemokine [C-C motif] receptor 4), TARC (thymus and activation regulated chemokine) et MDC (macrophage-derived chemokine), retrouvée par différents travaux. L’infiltrat lymphoplasmocytaire observé dans la MAG-4 est polyclonal, associant des lymphocytes T CD4+ et CD8+, des lymphocytes B et des plasmocytes IgG4. Il existe volontiers des centres germinatifs dans les tissus, qui semblent plus fréquents, plus nombreux et de plus grande taille que ceux observés dans le syndrome de Gougerot-Sjögren. L’analyse des sous-populations lymphocytaires T retrouve un contingent important de lymphocytes T régulateurs au niveau tissulaire et dans le sang circulant, ce qui est très différent des pathologies auto-immunes. La présence de ces lymphocytes T régulateurs, associée à une production locale de TGF-″, (transforming growth factor ″) pourrait être en grande partie responsable de l’apparition d’une fibrose tissulaire abondante, l’une des caractéristiques histologiques de la maladie. Dans ce type de réaction inflammatoire, l’activation des lymphocytes B et la commutation isotypique vers les IgG4 sont induites par l’IL-4 et l’IL-10. La corrélation entre les taux d’IL-4, d’IL-10, l’expression de FoxP3 et le ratio IgG4/IgG au niveau tissulaire chez les malades renforcent cette hypothèse. Un mécanisme T-indépendant a également été suggéré par un récent travail, dans lequel la production d’IgG4 serait dépendante de BAFF, produite par les monocytes ou les basophiles après stimulation des récepteurs Toll-like et NOD-like. BAFF (B-cell activating factor), cytokine importante pour la différenciation, la survie et la production d’Ig par les lymphocytes B, est retrouvée à des taux élevés dans le sang et dans les tissus des patients. L’inflammation tissulaire de la MAG-4 est donc mieux caractérisée, mais le ou les agents déclenchants restent inconnus.
Les IgG4 elles-mêmes ne semblent pas avoir de rôle dans la physiopathologie de la maladie. En effet les IgG4 ont des propriétés singulières, car elles ne fixent pas le complément par leur fragment Fc et auraient plutôt un rôle anti-inflammatoire, lié à leur capacité de devenir des anticorps bispécifiques par échange des chaînes lourdes et fragments Fab. Les IgG4 peuvent former des « pseudo-facteurs rhumatoïdes » par des inter-actions Fc-Fc dont le rôle pathogène n’est pas démontré.
Critères de diagnostic
Il faut distinguer les critères diagnostiques, spécifiques des différentes atteintes d’organes de la MAG-4, des critères diagnostiques « généraux » de la MAG-4. Ces critères ont été établis à partir de cohortes de patients, en comparaison à d’autres pathologies, soit spécifiques d’organes, soit auto-immunes comme le syndrome de -Gougerot-Sjögren.
Critères spécifiques
Il existe des critères spécifiques pour les pancréatites associées aux IgG4, les néphrites tubulo-interstitielle associées aux IgG4 et la sialadénite associée aux IgG4. Les critères de la pancréatite associée aux IgG4 permettent de la différencier de la pancréatite auto-immune de type 2 et du cancer du pancréas. Les critères dits « HISORt » reposent sur des critères histologiques, radiologiques, sérologiques, cliniques (atteintes extrapancréatiques) et de réponse au traitement (corticothérapie). Ces critères ont été remplacés plus récemment par un consensus international. Les critères de la néphrite tubulo-interstitielle associée aux IgG4 ont été proposés par des équipes japonaises et américaines. Des critères diagnostiques spécifiques de la sialadénite associée aux IgG4 (syndrome de Mikulicz) ont également été proposés par Masaki et al. en 2010 et permettent de la distinguer du syndrome de Gougerot-Sjögren.
Critères généraux
Ces critères ont été proposés par les équipes japonaises avec pour objectif de simplifier et de faciliter le diagnostic de la maladie pour les non-spécialistes. Il s’agit des comprehensive diagnostic criteria (ou CDC) [10], qui peuvent s’appliquer à tous les types d’atteinte et qui permettent d’orienter le clinicien vers un diagnostic possible, probable ou défini sur la base de la présence d’un critère morphologique (clinique ou radiologique), d’un critère biologique et d’un critère histologique (Tableau S03-P01-C27-I). Le critère morphologique correspond à l’augmentation de volume d’un ou de plusieurs organes, secondaire à l’infiltrat inflammatoire, pouvant prendre parfois des aspects pseudo-tumoraux. Il peut être évident cliniquement, mais doit le plus souvent être recherché par imagerie comme par exemple au niveau du pancréas. La tomographie par émission de positons (TEP) peut être utile pour dépister les différents organes atteints par la maladie [4].
Le critère biologique est unique et correspond à l’élévation des IgG4 sériques au-delà d’un seuil fixé à 1,35 g/l (ou 135 mg/dl), qui correspond à celui proposé initialement par H. Hamano et al. [5]. La technique de mesure n’est pas prise en compte. La sensibilité et la spécificité de ce dosage ont été évaluées dans différentes atteintes de la maladie, essentiellement au cours de l’atteinte pancréatique. Un seuil supérieur à 1,40 g/l est associé à une sensibilité de 76 % et une spécificité de 93 %, alors qu’un seuil supérieur à 2,8 g/l diminue la sensibilité à 53 % avec une spécificité de 99 % pour le diagnostic de pancréatite associée aux IgG4. La sensibilité est limitée par un pourcentage de patients, compris entre 10 et 20 % dans la majorité des séries, qui présentent des taux sériques d’IgG4 normaux [9]. Il peut s’agir dans certains cas de faux négatifs liés à un effet « prozone » (une dilution préalable du sérum permet de corriger cette erreur de mesure). La deuxième limite concerne la spécificité de ce dosage. De nombreux travaux ont montré qu’une élévation des IgG4 sériques au-delà du seuil proposé pouvait s’observer dans de nombreuses autres situations pathologiques [3].
Le troisième et dernier critère est histologique. Il y a deux types de critères histologiques requis pour le diagnostic. Le premier repose sur l’analyse en histologie conventionnelle, avec la mise en évidence d’une infiltration lymphocytaire et plasmocytaire polyclonale marquée, associée à une fibrose. Il peut s’y associer un infiltrat modéré à éosinophiles et des images de phlébites oblitérantes, mais ces éléments ne font pas partie des critères. La fibrose est habituellement très abondante, mais n’est pas retrouvée dans l’atteinte ganglionnaire. L’autre critère repose sur la mise en évidence d’une composante importante de plasmocytes IgG4+ au sein de l’infiltrat mononucléé en immunohistologie. Leur présence doit être quantifiée par le ratio plasmocytes IgG4+/IgG+ supérieur à 40 % et par le décompte dans les zones d’infiltrat cellulaire dense des plasmocytes IgG4+ qui doivent plus de 10 par champ à fort grossissement. Cette quantification est un critère important, car la présence de plasmocytes IgG4+ tissulaires n’est pas totalement spécifique de la maladie. Un consensus a été publié par un groupe international d’anatomopathologistes, définissant des seuils selon le type de tissu analysé [1].
Diagnostic différentiel
Le diagnostic de MAG-4 doit rester actuellement un diagnostic d’élimination, notamment du fait des limites de la spécificité de l’élévation des IgG4 sériques, voire de certaines caractéristiques histologiques. La présentation clinique ou radiologique pseudo-tumorale, souvent présente au diagnostic, doit faire éliminer une éventuelle pathologie néoplasique. Ainsi, le cancer du pancréas reste le principal diagnostic différentiel à éliminer devant une atteinte pancréatique. Une pathologie lymphomateuse doit également être éliminée, en particulier devant un tableau de polyadénopathies très souvent présent au diagnostic [2]. Notons que des observations de cancers solides et de lymphomes (en particulier de MALT ou mucosa-associated lymphoid tissue) ont été rapportées chez des patients atteints de MAG-4, sans qu’un lien de causalité formel soit retenu à ce jour.
1. Examen clinique : hypertrophie localisée ou diffuse au sein d’un ou de plusieurs organes classiquement atteints au cours de la MAG-4 (ce critère peut également reposer sur des données d’imagerie conventionnelle) |
2. Biologie : élévation des IgG4 sériques (ε 135 mg/dl ou 1,35 g/l) |
3. Histologie montrant : – infiltration lymphocytaire et plasmocytaire polyclonale marquée avec fibrose – infiltration par des plasmocytes IgG4+ : ratio plasmocytes IgG4+/IgG+ > 40 % – et/ou > 10 plasmocytes IgG4+ par champ à fort grossissement |
MAG-4 définie = (1) + (2) + (3) MAG-4 probable = (1) + (3) MAG-4 possible = (1) + (2) |
Il faut, dans tous les cas, éliminer, en particulier au niveau histologique, les principaux diagnostics différentiels de la MAG-4 : tumeur maligne (cancer, lymphome) et les tableaux cliniques proches : syndrome de Gougerot-Sjögren, cholangite sclérosante primitive, maladie de Castleman, fibrose rétropéritonéale secondaire, granulomatose avec polyangéite (Wegener), sarcoïdose, granulomatose éosinophilique avec polyangéite (Churg-Strauss) |
Le diagnostic différentiel se fait également avec certaines pathologies systémiques comme le syndrome de Gougerot-Sjögren, dont l’atteinte des glandes salivaires et lacrymales partage certaines similitudes cliniques avec celle de la MAG-4. Cependant, les anticorps anti-SS-A (Ro) et anti-SS-B (La) sont négatifs au cours de la MAG-4, et il existe des différences épidémiologiques, cliniques et surtout histologiques entre ces deux entités.
Une maladie de Castleman multicentrique peut également constituer un diagnostic différentiel difficile en cas d’atteinte ganglionnaire, d’autant qu’il existe des similitudes histologiques entre les deux pathologies. Cependant, les signes généraux sont souvent moins marqués au cours de la MAG-4, avec généralement un syndrome inflammatoire absent ou modéré, et l’absence d’élévation de l’IL-6 sérique.
Les autres diagnostics différentiels à évoquer sont la sarcoïdose, la granulomatose avec polyangéite (Wegener) (il n’existe pas de lésion granulomateuse ou de vascularite au niveau histologique au cours de la MAG-4), la granulomatose éosinophilique avec polyangéite (Churg-Strauss), la cholangite sclérosante primitive (fréquemment associée aux maladies inflammatoires de l’intestin), la maladie de Rosai-Dorfman ou encore les fibroses rétropéritonéales primitives ou secondaires à d’autres causes.
Manifestations cliniques
Signes généraux
Le tableau clinique est généralement d’installation progressive, et rarement associé à une fièvre très élevée, même si des tableaux systémiques bruyants sont rapportés à la phase initiale de certaines atteintes d’organes. Des signes généraux à type d’asthénie ou encore d’amaigrissement sont rapportés chez une proportion significative de patients (respectivement 56 et 44 % dans la cohorte nationale française) [2].
Signes liés aux atteintes d’organes
Les principales atteintes d’organes rapportées au cours de la MAG-4 sont présentées dans le Tableau S03-P01-C27-II. Le nombre de celles-ci augmente rapidement dans la littérature médicale, même si l’intégration à la MAG-4 de certaines d’entre elles reste encore discutée (par exemple, l’atteinte cutanée).
Pancréatite associée aux IgG4 (ou pancréatite « auto-immune » de type 1)
L’atteinte pancréatique est la première à avoir été rapportée et constitue l’atteinte la plus documentée dans la littérature. Le tableau clinique est « pseudo-tumoral » avec l’apparition progressive d’un ictère (63 % des cas), parfois associé à des douleurs abdominales (27 % des cas) et à un amaigrissement [6]. Une insuffisance pancréatique endocrine (diabète) et/ou exocrine peut survenir au cours de l’évolution chez 30 à 40 % des patients. L’imagerie (tomodensitométrie et/ou IRM) retrouve classiquement une hypertrophie pancréatique focale ou diffuse, avec volontiers un « anneau » hypodense péripancréatique. L’aspect du canal pancréatique en cholangio-pancréatographie rétrograde endoscopique est caractéristique avec rétrécissements multiples ou diffus, typiquement sans dilatation en amont. L’histologie est celle d’une pancréatite sclérosante lymphoplasmocytaire, avec prédominance de plasmocytes IgG4+.
Cholangite sclérosante associée aux IgG4
La cholangite sclérosante associée aux IgG4 est volontiers associée à la pancréatite auto-immune de type 1, même si elle peut se manifester en l’absence de toute atteinte pancréatique, rendant le diagnostic différentiel alors difficile avec une cholangite sclérosante primitive ou un cholangiocarcinome. La présentation est celle d’une cholestase avec un éventuel ictère d’aggravation progressive. Des critères diagnostiques spécifiques de cette atteinte d’organe ont été proposés, associant des anomalies radiologiques biliaires (rétrécissements segmentaires ou diffus des voies biliaires intra- et/ou extrahépatiques, avec épaississement pariétal de celles-ci), une élévation des IgG4 sériques, l’existence d’autres atteintes d’organes extrabiliaires et une histologie caractéristique. Contrairement à la cholangite sclérosante primitive, l’association à une maladie inflammatoire chronique de l’intestin n’est pas habituelle.
Sialadénite associée aux IgG4
L’atteinte des glandes salivaires se présente le plus souvent par l’hypertrophie d’une ou de plusieurs glandes salivaires (parotides et/ou sous-mandibulaires) (Figure S3-P1-C27-1a). Lorsque celle-ci est symétrique et touche au moins deux paires de ces glandes salivaires et/ou s’associe à une hypertrophie des glandes lacrymales, on parle de syndrome de Mikulicz. Un tableau de sialadénite sclérosante chronique, touchant habituellement les glandes sous-mandibulaires, réalise le tableau de tumeur de Küttner. La survenue d’un syndrome sec est moins fréquente qu’au cours du syndrome de Gougerot-Sjögren, probablement en raison de l’absence d’atteinte lympho-épithéliale. Parmi les autres éléments permettant de la distinguer d’un syndrome de Gougerot-Sjögren, on notera la moindre prévalence féminine, l’absence d’anticorps anti-SS-A (Ro) ou anti-SS-B (La), l’élévation des IgG4 sériques et la présence de plasmocytes IgG4+ au niveau tissulaire.
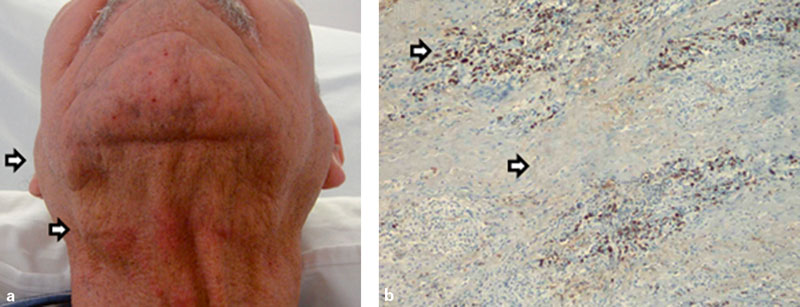
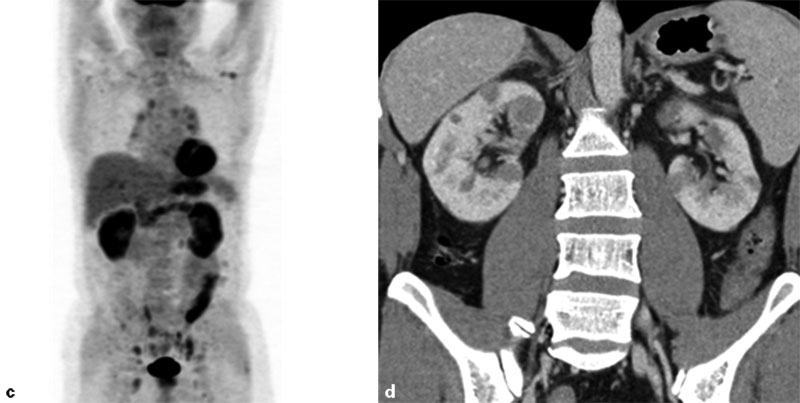
Exemples d’atteintes d’organes au cours de la maladie associée aux IgG4. a) Sialadénite associée aux IgG4 : patient présentant une tuméfaction bilatérale des glandes parotides et sous-maxillaires (flèches). b) Immunohistochimie avec un anticorps anti-IgG4 montrant une composante importante de plasmocytes IgG4+ dans les zones inflammatoires (flèche). Noter une fibrose marquée (flèche), caractéristique de la maladie. c) Tomographie par émission de positons réalisée dans le bilan d’extension d’une rechute de la maladie associée aux IgG4, montrant un hypermétabolisme spécifique des deux reins, du pancréas et d’adénopathies sus- et sous-diaphragmatiques. a) Tomodensitométrie chez un patient présentant une atteinte pseudo-tumorale rénale bilatérale.
Organes | Type d’atteinte (syndrome) | Manifestations cliniques |
|---|---|---|
Pancréas | Pancréatite sclérosante lymphoplasmocytaire (ou pancréatite auto-immune de type 1) | Ictère, douleurs abdominales, diabète, insuffisance pancréatique exocrine |
Voies biliaires/foie | Cholangite associée aux IgG4 Pseudo-tumeur inflammatoire hépatique | Ictère |
Glandes salivaires | Sialadénite associée aux IgG4 (syndrome de Mikulicz) Tumeur de Küttner | Hypertrophie parotidiennes et/ou sous-mandibulaire Syndrome sec, rare mais possible |
Glandes lacrymales | Dacryoadénite associée aux IgG4 (syndrome de Mikulicz) Pseudo-tumeur inflammatoire orbitaire/inflammation orbitaire idiopathique | Hypertrophie des glandes lacrymales, volontiers bilatérale |
Rétropéritoine | Fibrose rétropéritonéale (maladie d’Ormond) | Douleurs abdominales/lombaires, compression veineuse/urétérale |
Aorte | Aortite thoracique associée aux IgG4 Anévrysme aortique inflammatoire Péri-aortite | Asymptomatique le plus souvent, risque potentiel d’évolution anévrysmale, voire dissection |
Reins | Néphrite tubulo-interstitielle (très rarement, glomérulonéphrite extramembraneuse) | Asymptomatique le plus souvent, anomalies morphologiques rénales Insuffisance rénale aiguë ou chronique |
Ganglions | Adénopathies associées aux IgG4 | Polyadénopathies systémiques ou régionales, absence de signes généraux |
Poumon | Pseudo-tumeurs inflammatoires pulmonaires Pneumonie interstitielle | Découverte radiologique, toux/dyspnée/douleur |
Thyroïde | Thyroïdite fibrosante de Riedel Thyroïdite de Hashimoto dans son variant fibrosant, discuté | Goitre fibreux, compressif |
Hypophyse | Hypophysite associée aux IgG4 | Hypopituitarisme, diabète insipide, anomalies IRM |
Méninges | Pachyméningite associée Pseudo-tumeur inflammatoire méningées | Signes neurologiques selon la localisation |
Prostate | Prostatite associée aux IgG4 | Asymptomatique ou signes fonctionnels urinaires |
Dacryoadénite associée aux IgG4 et pseudo-tumeurs inflammatoires orbitaires
L’hypertrophie des glandes lacrymales, fréquemment bilatérale, peut être isolée, ou associée à l’atteinte des glandes salivaires (syndrome de Mikulicz). Outre l’atteinte des glandes lacrymales, une partie des tableaux d’« inflammation orbitaire idiopathique » peut être intégrée à la MAG-4, en raison d’une histologie caractéristique. La survenue de lymphomes, en particulier de type MALT, chez des patients suivis pour une dacryo-adénite associée aux IgG4 a été rapportée.
Fibrose rétropéritonéale associée aux IgG4
La présentation clinique est représentée le plus souvent par des douleurs abdominales et/ou lombaires d’apparition progressive, parfois associées à des signes généraux (fièvre, amaigrissement). L’imagerie (tomodensitométrie, IRM) permet de confirmer le diagnostic et de rechercher des complications (hydronéphrose par engainement urétéral, compressions veineuses). Selon les séries, la MAG-4 pourrait représenter jusqu’à 50 % des fibroses rétropéritonéales dites « idiopathiques ».
Maladie rénale associée aux IgG4
Cette atteinte est le plus souvent asymptomatique. Elle est fréquemment découverte devant des anomalies morphologiques rénales en imagerie (plus de 50 % des patients avec atteinte rénale : infiltration rénale, lésions nodulaires ou hypertrophie rénale, le plus souvent bilatérales), des anomalies du sédiment urinaire et/ou l’existence d’une insuffisance rénale aiguë ou chronique (jusqu’à 70 % des patients avec atteinte rénale). L’histologie retrouve, dans la très grande majorité des cas, une néphrite tubulo-interstitielle avec les caractéristiques typiques : infiltrat lymphoplasmocytaire, avec prédominance de plasmocytes IgG4+, et fibrose « storiforme ». Plus rarement, une atteinte glomérulaire de type glomérulonéphrite extramembraneuse a été rapportée. L’existence d’une hypocomplémentémie, d’une élévation importante des IgG4 sériques et d’une présentation multisystémique semble plus fréquente chez les patients avec atteinte rénale.
Adénopathies associées aux IgG4
La présence d’adénopathies, systémiques ou régionales d’une atteinte d’organe, est fréquente au cours de la MAG-4 (près de 70 % des patients dans la cohorte française) [2]. Il s’agit, le plus souvent, de tableaux de polyadénopathies, sans signes généraux associés. Cinq types histologiques possibles ont été individualisés (Castleman-like, hyperplasie folliculaire, expansion interfolliculaire, transformation progressive des centres germinatifs et pseudo-tumeur inflammatoire-like). La fibrose est habituellement absente dans l’atteinte ganglionnaire, et le seuil de plasmocytes IgG4+ requis est supérieur aux autres atteintes d’organes (> 100 par champ à fort grossissement, ratio > 40 %). Devant le caractère non spécifique de l’infiltrat plasmocytaire IgG4+, l’existence d’une atteinte ganglionnaire sans autre atteinte d’organe extraganglionnaire évocatrice est à prendre avec précaution.
Aortite associée aux IgG4
Atteinte le plus souvent asymptomatique, il existe cependant un risque évolutif potentiel, le diagnostic ayant été porté sur des pièces opératoires de patients atteints d’anévrysmes ou de dissections de l’aorte thoracique. L’aortite associée aux IgG4 rendrait compte de près de 9 % des cas d’aortites thoraciques. Il s’agit d’une aortite lymphoplasmocytaire, dont l’infiltrat inflammatoire touche principalement l’adventice, même si une atteinte de la media est possible. Lorsqu’une histologie est disponible, l’absence de granulome est un élément important du diagnostic différentiel. Un anévrysme inflammatoire de l’aorte abdominale (ou « péri-aortite ») constitue un autre mode de présentation de l’atteinte aortique.
Autres atteintes d’organes
Parmi les autres atteintes d’organes rapportées (voir Tableau S03-P01-C27-II), on pourra citer : des atteintes thoraciques (pseudo-tumeurs inflammatoires pulmonaires, pneumonies interstitielles, médiastinites fibrosantes, lésions pleurales fibro-inflammatoires) ; thyroïdiennes (thyroïdite fibrosante de Riedel) ; hypophysaires (hypophysite associée aux IgG4) ; méningées (pachyméningite associée aux IgG4 et pseudo-tumeurs inflammatoires méningées) ; ou encore urologiques (prostatite associée aux IgG4, mais également plus rarement atteintes urétérales ou testiculaires). L’intégration de certaines atteintes d’organes à la MAG-4 reste encore discutée : atteintes cutanées (pseudo-lymphomes cutanés), certaines atteintes thyroïdiennes (thyroïdite de Hashimoto dans son variant fibrosant), ou encore certaines atteintes ORL, en particulier nasales.
Signes biologiques
La première anomalie biologique rapportée chez les patients atteints de pancréatite auto-immune est la présence d’une hypergammaglobulinémie polyclonale à l’électrophorèse des protéines plasmatiques. Celle-ci est présente chez près de 75 % des patients atteints de MAG-4, son absence n’excluant pas le diagnostic.
Le marqueur biologique principal de la MAG-4 est l’élévation des IgG4 sériques, dont les limites en termes de sensibilité et de spécificité ont été décrites plus haut (voir « intertitre S3-P1-C27-N3-2 »). Une augmentation d’une ou de plusieurs autres sous-classes (IgG1 et/ou IgG2 en particulier) peut être présente chez certains patients, mais dans une proportion moindre que celle observée pour les IgG4 [2]. Une augmentation des chaînes légères libres a également été rapportée, avec une augmentation du rapport κ/λ chez certains patients.
Une hypocomplémentémie est observée chez près de 25 % des patients. Celle-ci porte sur le C3, le C4 et/ou le CH50. Elle semble concerner préférentiellement les patients présentant une atteinte rénale. Le mécanisme de cette hypocomplémentémie reste mal connu, les IgG4 n’activant pas la voie classique du complément.
Un syndrome inflammatoire biologique peut être présent chez 10 à 15 % des patients [2], habituellement modéré.
Enfin, les anticorps antinucléaires peuvent être positifs, habituellement à faible taux et sans spécificité. Les anticorps anti-SSA et anti-SSB sont en revanche négatifs chez ces patients, élément important pour le diagnostic différentiel avec le syndrome de Gougerot-Sjögren.
Traitement
Aucune étude contrôlée randomisée n’est actuellement disponible dans le cadre du traitement de la MAG-4. En dehors de quelques situations particulières (par exemple un tableau de polyadénopathies isolées, asymptomatiques), la mise en route d’un traitement paraît le plus souvent indiquée en raison du caractère fibrosant de la maladie. La corticothérapie constitue le traitement de première intention, avec une excellente cortico-sensibilité chez plus de 90 % des patients [2]. La réponse très favorable aux corticoïdes a d’ailleurs été proposée comme un critère diagnostique de la maladie. Le schéma d’administration et de décroissance de la corticothérapie n’est pas consensuel, et les données sont principalement issues de travaux sur la pancréatite auto-immune de type 1. Après un traitement d’attaque habituellement de 0,4 à 0,6 mg/kg/j, certaines équipes américaines proposent une décroissance rapide avec un arrêt à 11 semaines, alors que des équipes japonaises proposent un traitement d’entretien plus prolongé autour de 5 mg/j pendant 12 à 24 mois, voire 36 mois. Quel que soit le schéma, le taux de rechute est important à l’arrêt du traitement. Au cours des pancréatites auto-immunes de type 1, le taux de rechutes est de 32 % à 6 mois, 56 % à 1 an et 92 % à 3 ans [7]. Un traitement immunosuppresseur de deuxième ligne est donc souvent utilisé, soit à visée d’épargne cortisonique, soit pour limiter les rechutes. L’azathioprine constitue la molécule la plus utilisée dans le cadre de la pancréatite auto-immune de type 1, même si l’utilisation d’autres traitements a été rapportée (mycophénolate mofétyl, cyclophosphamide ou encore bortézomib). De bonnes réponses ont également été rapportées avec l’utilisation d’anticorps anti-CD20 (rituximab) chez des patients réfractaires ou présentant des effets secondaires aux traitements usuels, y compris pour certaines atteintes avec fibrose avancée.
Conclusion
Le concept de maladie associée aux IgG4 a permis de regrouper, autour de critères histologiques communs, de nombreuses entités syndromiques décrites de longue date et souvent associées. Le diagnostic doit être posé sur des critères stricts, notamment histologiques avec étude en immunohistochimie, après avoir éliminé les nombreux diagnostics différentiels. Le caractère systémique lié au grand nombre d’organes pouvant être atteints, de façon souvent concomitante ou au cours du temps, implique de nombreux spécialistes dans sa prise en charge. Ceci impose un bilan soigneux à la recherche des différentes atteintes chez ces patients. La maladie se caractérise par une grande cortico-sensibilité, mais celle-ci ne doit pas faire oublier le risque de séquelles liées au caractère fibrosant de l’affection. De plus, l’évolution chronique et la fréquence élevée des rechutes justifient un suivi régulier des patients, et l’utilisation éventuelle de secondes lignes thérapeutiques, dont les modalités doivent encore être précisées.
Bibliographie